
前言
保守的Ser/Thr蛋白激酶mTORC1是调节各种细胞合成代谢过程和途径的核心催化成分,它在溶酶体表面被激活。激活mTORC1需要两个必要条件:首先,mTORC1被V-ATPase-Ragulator-Rag复合物引入到溶酶体表面,以响应氨基酸和其他营养物质;其次,mTORC1被溶酶体结合的GTP酶Rheb激活,以应对生长因子-PI3K-AKT-TSC1/2信号途径。mTORC1最著名的底物是两种调节蛋白合成的核糖体蛋白S6激酶(S6K)和eIF4E结合蛋白(4EBP)。此外,mTORC1还可以通过SREBP和PPAR-γ调节脂质合成和脂肪组织分化。
最近一些研究表明,溶酶体不仅控制mTORC1的活化,而且还接收来自mTORC1的信号,以调节许多重要的生物过程。一个关键问题是,一旦mTORC1在溶酶体表面被激活,它是否磷酸化,并通过局部溶酶体蛋白发出信号。
本篇为2022年4月,由德克萨斯大学西南医学中心Yonghao Yu研究团队在Nature Communications(IF:17.674)期刊上发表了题为“Quantitative phosphoproteomic analyses identify STK11IP as a lysosome-specific substrate of mTORC1 that regulates lysosomal acidification”的研究成果,本文作者运用磷酸化蛋白组学、TMT标记定量蛋白质组学、非靶向代谢组学等组学方法,对溶酶体蛋白质组与mTORC1调节的磷酸化蛋白质组的交叉参考分析,将STK11IP鉴定为mTORC1的溶酶体特异性底物,随后对STK11IP进行敲除等研究,结果表明STK11IP磷酸化是mTORC1调节溶酶体酸化和自噬的一种机制,并指出STK11IP是改善自噬信号异常疾病的一个潜在的治疗靶点。

研究思路

研究结果
1.mTORC1调控溶酶体磷酸化蛋白质组的综合分析
因为蛋白质磷酸化的丰度低,所以细胞器水平的研究是个挑战。为了克服这个问题,作者开发一种方法。首先,使用质谱仪分别检测了溶酶体蛋白质组和总的mTORC1调控的磷酸化蛋白质组。然后,交叉引用了两个数据集,并用生物信息学提取了潜在的溶酶体特异性mTORC1底物。
使用免疫沉淀(IP)实验分离溶酶体(图1 a),运用蛋白质组实验从中鉴定出了231种蛋白质。GO分析表明,这些蛋白大多分布在液泡和溶酶体,富集到囊泡介导的转运、内吞作用和离子转运生物学过程,这都与溶酶体生物学功能有关。
使用基于还原二甲基化的定量质谱方法,检测了由胰岛素控制的肝脏磷酸化蛋白质组(在原代大鼠肝细胞中)及其关键的下游激酶,包括Akt,mTORC1和S6K。与溶酶体蛋白质组学数据对比,共鉴定出45种蛋白质(图1 b,c)。然后,从肝脏磷酸化蛋白质组学数据集中提取了作为潜在mTORC1底物的蛋白质,即雷帕霉素处理后磷酸化减少,S6K抑制剂处理后磷酸化不变的蛋白质。STK11IP也在溶酶体蛋白质组学实验中被鉴定出来(图1 b,c)。
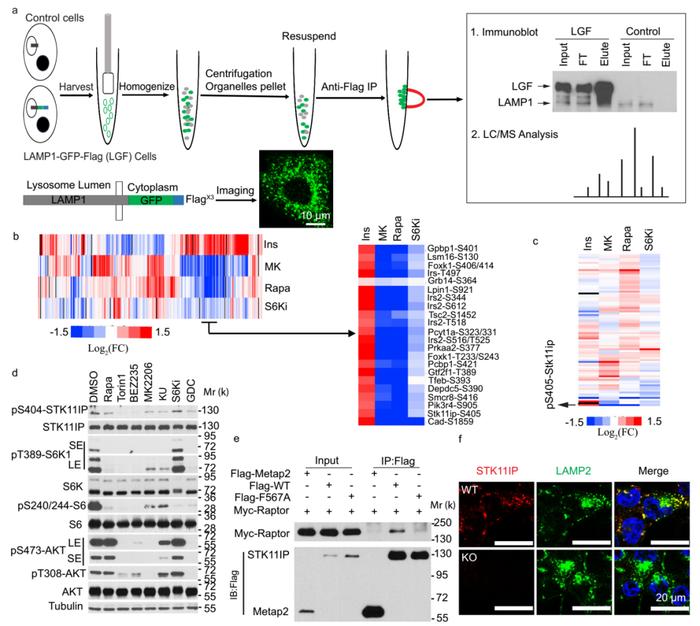
图1 mTORC1调节溶酶体磷酸化蛋白质组的综合分析
2. STK11IP是mTORC1的溶酶体特异性底物
STK11IP的磷酸化在雷帕霉素处理后显著降低,与已知的mTORC1磷酸化基序一致。实验发现STK11IP(pS404)的磷酸化模式与其他已知的mTORC1底物ULK1(pS757)相似,但与已知的S6K底物不同。这些结果表明STK11IP(pS404)可能是mTORC1的直接底物。
生成了针对STK11IP/pS404的磷酸化特异性抗体,发现pS404被雷帕霉素和mTOR激酶抑制剂有效抑制。但是S6K抑制剂对该磷酸化位点无效(图1 d)。对STK11IP序列的检查表明,该蛋白含有潜在的TOS基序,这个TOS基序(F567A)中关键残基(F567)的突变取消了STK11IP和Raptor之间的结合(图1 e)。这进一步证明了STK11IP是mTORC1的直接底物。免疫共染色显示,溶酶体中存在STK11IP(图1 f)。
3.STK11IP缺乏促进自噬
许多mTORC1底物通过反馈机制调节其活化,但是实验没有观察到对照细胞和STK11IP敲除(KO)原代小鼠胚胎成纤维细胞(MEF)细胞之间mTORC1活性有任何差异。使用CRISPR-Cas9系统生成STK11IP KO细胞,并与WT-、S404A-或S404D-STK11IP进行重组。然后进行了氨基酸饥饿实验,发现这些细胞中的mTORC1活性没有差异。
随后,研究了STK11IP是否可以介导mTORC1下游的某些生物过程,mTORC1可通过其下游靶蛋白(例如TFEB和ULK1)参与控制自噬。实验发现pS404-STK11IP的下调与自噬水平的上调同时发生,例如FBS或FBS/葡萄糖饥饿时LC3I到LC3II的转化率增加,还观察到pS404-STK11IP的下调与LC3的降解一起发生。这些结果表明,STK11IP可能作为mTORC1的下游靶标来调节自噬。
为了验证,使用CRISPR-Cas9敲除HEK293T细胞中的STK11IP。与对照相比,自噬体数的标志物LC3II在STK11IP KO细胞中显著增加(图2a,b)。此外,STK11IP KO细胞中的LC3点(内源性LC3)或GFP-LC3点(使用GFP-LC3稳定细胞)更多(图2c,d)。
自噬通量是自噬活性的可靠指标,于是作者进行了LC3周转率测定,来比较WT和STK11IP KO细胞之间的自噬通量。结果表明STK11IP KO使自噬通量增加了约两倍(图2 e,f)。使用另外一种自噬通量测量方法(荧光探针GFP-LC3-RFP),表明STK11IP KO细胞中的自噬通量增强(图2 k)。总的来说,与野生型细胞相比,STK11IP−/−细胞中自噬体和自溶酶体的形成均显著增加(图2 i)。因此,当STK11IP被删除时,自噬通量增加。
为了研究mTORC1介导的STK11IP磷酸化在自噬中的作用,作者在STK11IP KO细胞中表达了WT-,S404A-或S404D-STK11IP。总的结果表明,STK11IP在S404处的去磷酸化也促进了自噬通量(图2j-o)。
接下来,利用STK11IP KO小鼠来确定STK11IP是否调节体内自噬。自噬最有效的生理诱导剂之一是运动,使用WT和STK11IP KO同窝仔进行了耐力运动测试。发现与野生型小鼠相比,STK11IP KO小鼠在静息和耐力运动条件下在比目鱼肌中表现出更高水平的LC3II转化(图2 p,q)。总的来说,结果表明STK11IP KO促进体内自噬通量。
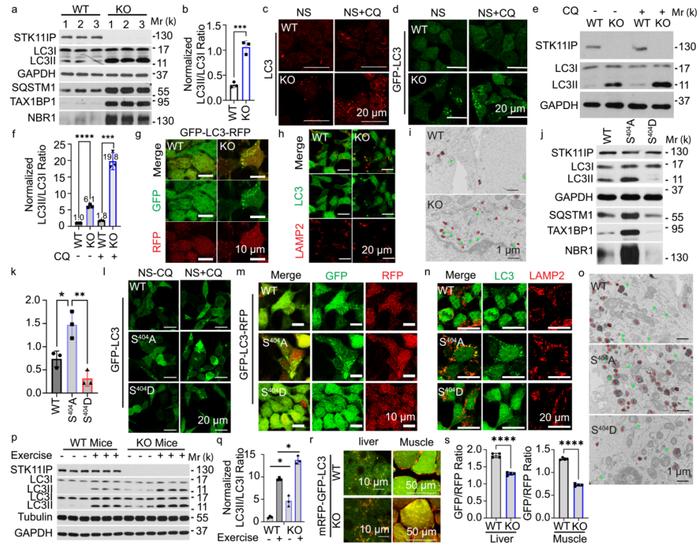
图2 STK11IP缺失促进自噬
4. STK11IP与V-ATP酶结合以调节溶酶体酸化
自噬是一个多步骤的过程,包括起始、自噬体形成和自噬体-溶酶体融合,没有发现对照组与STK11IP KO细胞的溶酶体生物发生和溶酶体形态水平有任何差异。观察到STK11IP KO细胞中的溶酶体相对于对照组的酸性更强(图3a)。此外,与表达WT-STK11IP或S404D-STK11IP突变体的细胞相比,用S404A-STK11IP突变体重建的STK11IP KO细胞也显示出更多的酸性溶酶体(图3 b)。使用比例探头来检测溶酶体pH值(基于黄色/蓝色信号的比率),表明STK11IP KO细胞和S404A-STK11IP表达细胞的pH值显著降低(图3 c,d)。使用pH 4.5纳米探针(pH阈值为4.5)时,只能在STK11IP KO和S404A-STK11IP重组细胞中检测到激活的TMR信号(图3 e,f),这与前面检测的结果一致。推测溶酶体通过质子泵送V型ATPase维持其pH梯度,所以研究了STK11IP是否对V-ATPase活性有影响。检测发现与对照细胞相比,从STK11IP KO和S404A-STK11IP细胞分离的溶酶体中V-ATPase的活性增强(图3 g,j)。
使用TMT标记定量蛋白质组技术对STK11IP WT和KO细胞进行了分析,发现STK11IP KO对V-ATPase各种组分的表达没有影响。然后使用TurboID系统来研究STK11IP是否可以与V-ATPase复合物相互作用(图3 k)。确定了V-ATP酶复合物的几种成分,包括ATP6V1B,ATP6V1A和ATP6V1H(图3 l)。使用co-IP进一步验证了STK11IP与V-ATPase复合物的各个亚基之间的相互作用。有趣的是,发现与WT蛋白相比,S404A-STK11IP突变体与V-ATPase组分(ATP6V1A和ATP6V1B)形成较弱的相互作用(图3 m)。这些数据表明,mTORC1介导的STK11IP在S404处的磷酸化对于其与V-ATPase复合物的相互作用至关重要。
综上,mTORC1信号传导调节STK11IP在S404处的磷酸化。STK11IP在S404处的去磷酸化削弱了其与V-ATPase的相互作用。STK11IP的去磷酸化导致V-ATP酶活性增加,溶酶体酸性增加,自溶酶体形成增强,自噬通量增强(图3o)。
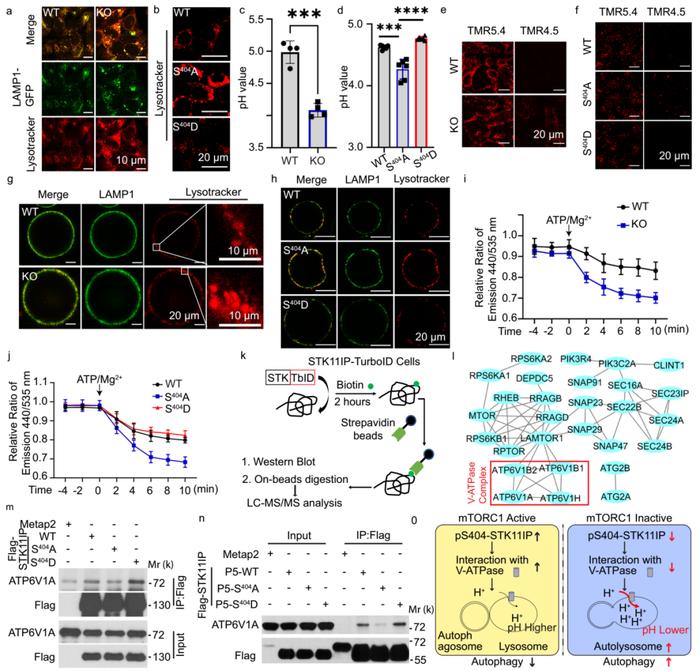
图3 STK11IP调节溶酶体酸化
5. STK11IP缺乏可预防空腹和MCD引起的脂肪肝
为了研究STK11IP在小鼠中的生理功能,测量了STK11IP在小鼠组织中的表达谱。qRT-PCR和免疫印迹分析结果显示,STK11IP在肝脏、白脂肪细胞组织(WAT)和胰腺中的表达较高。
在禁食36小时后,发现pS404-STK11IP的磷酸化水平降低。STK11IP KO小鼠显示出LC3I到LC3II转化率增加(图4 a)。这些结果进一步表明,STK11IP及其对pS404的磷酸化可能在这一过程中发挥作用。这种自噬的增强显著改善了空腹诱导的脂肪肝,表现为STK11IP KO小鼠中脂滴的减少(图4 b-d)。血清和肝脏中的TG和NEFA(非酯化脂肪酸)水平也随之降低(图4 e-g)。这些观察结果与之前的报告一致,即自噬在长期禁食期间促进脂质转化为葡萄糖。
为了进一步了解STK11IP KO后的代谢重塑,对从WT和STK11IP KO小鼠收获的血清非靶向代谢组学分析。分析显示糖生成、酮体代谢、脂肪酸β-氧化以及甜菜碱或肉碱代谢显著富集,这可以解释为什么长期饥饿后STK11IP KO小鼠的葡萄糖水平较高而TG水平较低(图e-h)。在饥饿时,自噬产生的脂肪酸和氨基酸可用于驱动糖异生和生酮。随着饥饿的继续,脂肪和肌肉组织的降解在向肝脏提供底物方面发挥着越来越大的作用,肝脏输出葡萄糖和酮体来喂养其他组织。
利用了由蛋氨酸-胆碱缺乏(MCD)饮食诱导的NASH模型。两周后,与WT小鼠相比,STK11IP KO小鼠的体重下降相似,然而,与对照细胞相比,STK11IP KO小鼠的肝脏显示出LC3II / LC3I比率增加(图4 j)。STK11IP KO小鼠表现出肝脂肪变性降低,如大囊泡脂滴减少所示(图4 k,l)和血清和肝组织中TG水平的下调(图4 m,n)。

图4 STK11IP缺乏症可保护小鼠免受代谢紊乱的侵害
研究讨论
总的来说,使用磷酸化蛋白组+常规定量蛋白质组学等一系列研究方法,可以将STK11IP鉴定为mTORC1的新型溶酶体特异性底物。STK11IP的敲除导致自噬通量的强劲增加。STK11IP在Ser处(404)的去磷酸化抑制STK11IP作为自噬抑制剂的作用。在机制上,STK11IP与V-ATPase结合,并调节V-ATPase的活性。敲除STK11IP可保护小鼠免受空腹或蛋氨酸/胆碱缺乏饮食(MCD)诱导的脂肪肝。因此,研究表明,STK11IP磷酸化是mTORC1调节溶酶体酸化和自噬的一种机制,并指出STK11IP是改善具有异常自噬信号传导的疾病的潜在治疗靶点。
小鹿推荐
通过溶酶体蛋白质组与mTORC1调节的磷酸化蛋白质组的交叉参考分析,将STK11IP鉴定为mTORC1的溶酶体特异性底物。通过对STK11IP的研究,发现它能调节溶酶体酸化和自噬溶酶体的形成。这为靶向STK11IP治疗具有异常自噬信号传导的疾病提供了基础。
详细技术请访问鹿明生物官网
⬇
百度搜索鹿明生物(lumingbio)
⬇
了解更多多组学技术
(磷酸化蛋白组学、TMT标记定量蛋白质组学、非靶向代谢组学)
猜你还想看
1,新品速递 | 高深度超微量蛋白质组学-切片类样本
2,技术前沿 | 最新深度报道!癌症空间代谢组学策略,“鉴往知来”找准癌症研究新方向~
3,Ann Neurol | 神经科学一区TOP期刊,代谢组学为鉴别急性缺血性卒中与拟卒中提供相关诊断价值
4,十月年度最佳 | 恭喜鹿明生物蛋白代谢30篇项目文章发表,总IF:230+
本文系鹿明生物原创
转载请注明本文转自鹿明生物


